Click on the author name to see the abstract.
| Chair: Colin J. Lambert | |
| 09:00-09:35 | Roche |
| 09:35-10:00 | Trioni |
| 10:00-10:25 | Del-Castillo |
| 10:25-11:00 | Coffee break |
| Chair: Alessandro Pecchia | |
| 11:00-11:35 | Segal |
| 11:35-12:10 | Nikolic |
| 12:10-12:35 | Avriller |
| 12:35-13:00 | Popescu |
| 13:00- | Free afternoon |
Invited talk
Real space computational approaches for exploring charge/spin/thermal transport in materials with billions of atoms
1Catalan Institute of Nanoscience Nanotechnology, Campus UAB, Bellaterra, Spain
2ICREA, Institució Catalana de Recerca i Estudis Avancats, Spain
I will overview two decades of development of real space developments of quantum transport methodologies which have allowed simulations of bulk charge/thermal and spin transport in model systems of remarkable complexity, including all sort of disorder (static and vibrational), varying dimensionality and system sizes reaching the experimental and technology relevant scales (for technical details read [1]). After some introduction of the scope of those methods, I will focus on most recent developments on quantum Hall (spin) conductivities which have allowed the exploration of unprecedented transport features in disordered Dirac Matter, including our recent exploration of spin dynamics in graphene and topological insulators which demonstrate the brute force capability of such methods as unrivaled tools for new fundamental discoveries [2,3].
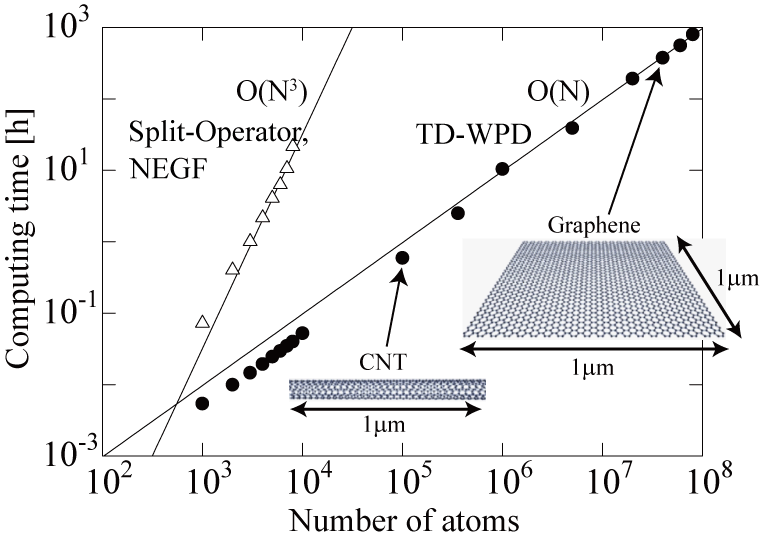
Figure 1: The unrivalled scaling behavior of real space quantum transport methodologies.
[1] L. E. F. Foa Torres, S. Roche, and J. C. Charlier, Introduction to Graphene-Based Nanomaterials: From Electronic Structure to Quantum Transport (Cambridge University Press, Cambridge, 2014).
[2] F. Ortmann and S. Roche, Phys. Rev. Lett 110, 086602 (2013). F. Ortmann, N. Leconte, and S. Roche, Phys. Rev B 91, 165117 (2015). N. Leconte, F. Ortmann, A. Cresti, and S. Roche, Phys. Rev B 93, 115404 (2016).
[3] D. Van Tuan et al., Nature Physics 10, 857–863 (2014); D. Van Tuan et al., Scientific Reports 6, 21046 (2016); A.W. Cummings and S. Roche, Phys. Rev. Lett. 116, 086602 (2016). A. Cresti, D. Van Tuan, D. Soriano, A. W. Cummings, S. Roche, Phys. Rev. Lett. 113, 246603 (2014). D. Van Tuan and S. Roche, Phys. Rev. Lett. 116, 106601 (2016). D. Van Tuan, J. M. Marmolejo-Tejada, X. Waintal, B.K. Nikolic, and S. Roche, arXiv:1603.03870.
Contributed talk
Spin-filtering in graphene junctions with Ti and Co adsorbates
1Physics Department, Catholic University of "Sacro Cuore", v. dei Musei 41, Brescia, Italy
2CNR, National Research Council of Italy, ISTM, via Golgi 19, Milan, Italy
3Chemistry Department, University of Milan, via Golgi 19, Milan, Italy
Organic materials have recently demonstrated their potentiality for applications in molecular spintronics due to their electronic and magnetic properties that, for some aspects, differentiate from those of the conventional inorganic metals and semiconductors. Carbon-based structures have been investigated as possible spintronics components [1,2], exploiting the possibility of inducing and controlling magnetism in graphene. The adsorption of transition metals (TM) is one of the most appealing techniques [3,4] because it can turn graphene into a half-metal [5].
We will analyze the spin-dependent current and spin-filtering efficiency of a graphene sheet adsorbed with TM atoms (Ti and Co), from first principles. Our calculation, exploiting the Non-Equilibrium Green's Function (NEGF) formalism implemented in TranSIESTA [6], shows that the adsorption of transition metal atoms on graphene induces the opening of a gap in the transmission function in one spin channel only. Charge carriers flowing through the nanojunction in Ti@graphene and Co@graphene belong almost entirely to the minority and majority spin component, respectively. This gives rise to a spin polarization of the current of about 100%.
Also the analysis of the non equilibrium electron density distribution indicates that a spin separation between the left and the right portions of the device occurs. This strong spin asymmetry suggests that transition metal adatoms have the potential of turning graphene into an efficient spin-filtering device.
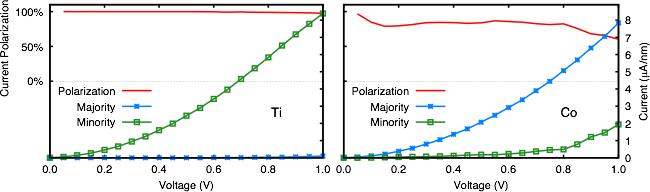
Figure 1: Electronic current and its spin polarization in Ti@graphene (left) and Co@graphene (right).
[1] J. Guo, J. D. Gunlycke, C. T. White, Appl. Phys. Lett. 92, 163109 (2008).
[2] K. Tsukagoshi, B. W. Alphenaar, H. Ago, Nature 401, 572 (1999).
[3] K. T. Chan, J. B. Neaton, M. L. Cohen, Phys. Rev. B 77, 195434 (2008).
[4] M. Manadè, F. Viňes, F. Illas, Carbon 95, 525 (2015).
[5] E. del Castillo, F. Cargnoni, S. Achilli, G. Tantardini, M. I. Trioni, Surf. Sci. 634, 62 (2015).
Contributed talk
Electron transport in a Fe-Porphyrin/Graphene junction
1Chemistry Department, University of Milan, via Golgi 19, Milan, Italy
2CNR, National Research Council of Italy, ISTM, via Golgi 19, Milan, Italy
A relatively new field of investigation is molecular spintronics, in which magnetic molecular junctions are used as spin transport channels [1]. Both experiments and theoretical works suggest that organic materials can offer similar and perhaps superior performances in making spin-devices than the more conventional inorganic metals and semiconductors [2]. Among organic materials, porphyrins are considered promising candidates because they offer a variety of desirable features such as highly conjugated structure, rigid planar geometry and good chemical stability [3].
In this work we study by first principles the transport properties of a magnetic molecular junction consisting of Fe-porphyrin molecule connected with two semi-infinite graphene electrodes. The calculations were performed using the TranSIESTA code [4], which combines the non-equilibrium Green's function (NEGF) technique with DFT. We find that the localized Fe states do not produce relevant effects on the current which in fact displays only a slight polarization.
We further investigate the electron transport through the same molecular junction contacted with boron and nitrogen doped graphene electrodes. The presence of the dopants leads to a non negligible density of states around the Fermi level allowing the hybridization between iron and carbon states. In the case of B-doped electrodes a current polarization is observed, while in the N-doped case a Negative Differential Resistance effect can be pointed out. With differently doped electrodes, one with boron and the other with nitrogen, the junction displays a partial rectification behavior.
Since the electronic properties of the metal atom can be modulated by the adsorption of a gas molecule, we study the effect of a gas molecule adsorption on the charge transport and we observe a quenching of the current polarization in the B-doped system.
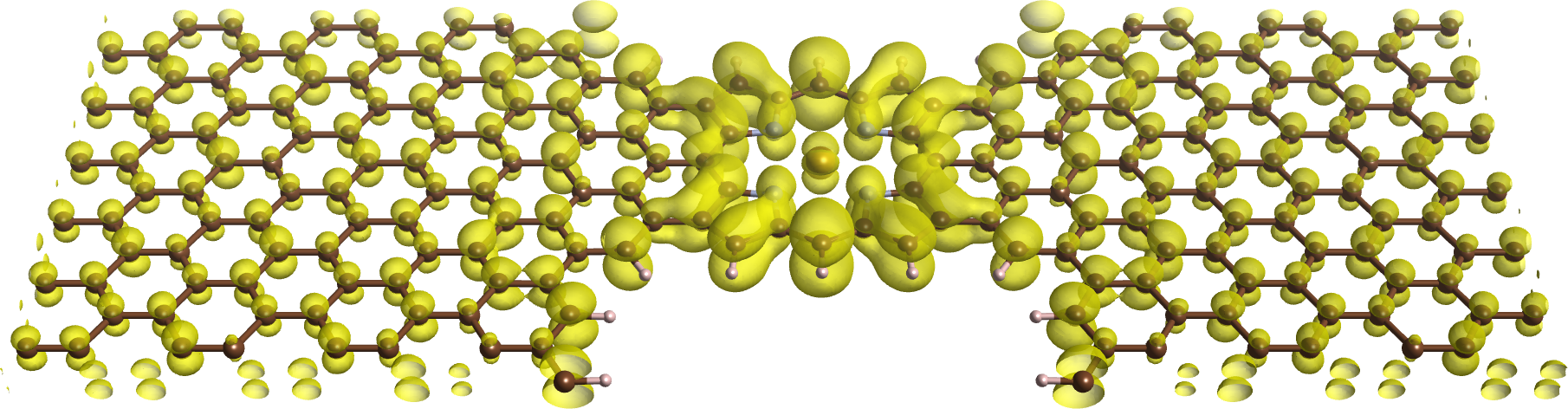
Figure 1: Local density of states of a molecular hybridized state of the nanojunction.
[1] A. R. Rocha et al., Nature Mater. 5, 335 (2005).
[2] S. Sanvito, Nature Mater. 6, 803 (2007).
[3] S. U. Lee et al., Small 7, 962 (2008).
[4] M. Brandbyge et al., Phys. Rev. B 65, 165401 (2002).
Invited talk
Buttiker's probes in molecular electronics: Applications to charge and heat transport
Department of Chemistry, University of Toronto, 80 Saint George St., Toronto, Ontario, Canada M5S 3H6
The role of incoherent (elastic, inelastic) scattering effects on charge and heat conduction in molecules is explored using the phenomenological Buttiker's probe method. In the past 30 years this easy-to-implement approach has been extensively employed to research inelastic effects in the transport behavior of mesoscopic devices, yet limited to the linear response regime. Our recent work has been focused on extending this method to far-from-equilibrium applications. In a series of recent papers we showed that the probe technique can reliably mimic incoherent charge transfer phenomena under finite voltage biases [1-3], as well as vibrational heat conduction under large temperature differences [4-5].
Concerning incoherent electronic conduction, I will begin by defining two types of constructions, the dephasing and voltage probes. I will then show that these probes can properly reproduce the tunneling-to-hopping crossover in conductance at low applied bias [1]. At high bias, we will identify signatures of elastic and inelastic scattering processes in the electrical current [3], use Buttiker's probes to examine the robustness of tunneling diodes [2], and propose mechanisms for environmentally-induced diodes [3]. If time allows, I will discuss applications of Buttiker's probes in studies of vibrational heat transfer through molecules, placed under high temperature differences. I will particularly demonstrate the utility of the method in materializing nonlinear function, e.g., thermal diodes operating in the quantum regime [4-5].
[1] M. Kilgour and D. Segal, J. Chem. Phys. 143, 024111 (2015).
[2] M. Kilgour and D. Segal, J. Phys. Chem. C, 119, 25291 (2015).
[3] M. Kilgour and D. Segal, J. Chem. Phys. 144, 124107 (2016).
[4] D. Segal, Phys. Rev. E 79, 012103 (2009).
[5] M. Bandyopadhyay and D. Segal, Phys. Rev. E 84, 011151 (2011).
Invited talk
Diagrammatic and resummation algorithms for electron-phonon and electron-magnon interacting systems in nanojunctions far from equilibrium
Department of Physics & Astronomy, University of Delaware, Newark, DE 19716, U.S.A.
The nanoelectronic and spintronic devices based on single-molecule or magnetic tunnel junctions are typically operated by finite bias voltage which can lead to highly nonequilibrium states of electrons and bosons (such as phonons or magnons). While nonequilibrium Green function (NEGF) formalism offers a rigorous theoretical and computational framework to describe the effect of their mutual interaction on charge and spin currents underlying the device functionality, the computational complexity of standard self-consistent diagrammatic many-body perturbation scheme for NEGFs has restricted simulations to very small junctions (containing few tens of atomic orbitals). Furthermore, magnons have small bandwidth meaning that weak interactions felt by electrons turns out to be strongly coupled regime for magnons, thereby requiring higher order diagrams in the expansion of magnonic NEGF. On the other hand, as the interactions strength increases, self-consistent diagrammatic series often converges to the unphysical branch. This talk will overview our recent NEGF-based approach [1] to nonequilibrium electron-magnon systems in magnetic tunnel junctions, where magnons become quasiparticles dressed by the cloud of virtual electron-hole pairs while electrons scattering of low frequency magnons experience anomaly in their current-voltage characteristics at small bias voltage. I will then use an example of a single molecule nanojunction to show how self-consistent diagrammatic series for NEGFs describing electron-phonon (or electron-magnon) inelastic scattering can be evaded by evaluating only fourth-order bare diagrams [2] and subsequently performing very recently proposed [3] hypergeometric resummation that preserves conservation laws and can make possible simulations of electron-boson coupled systems in devices containing very large number of atoms.
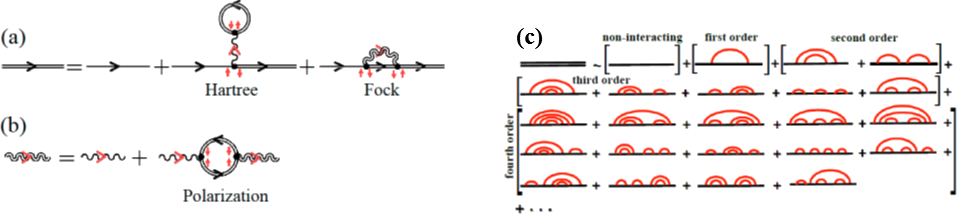
Figure 1: Example of self-consistent diagrammatic series for (a) electronic NEGF or (b) magnonic NEGF in nonequilibrium electron-magnon system, as well as fourth-order bare diagrammatic series for NEGF of electrons interacting with phonons which is used as an input for hypergeometric resummation.
[1] F. Mahfouzi and B. K. Nikolić, Phys. Rev. B 90, 045115 (2014).
[2] H. Mera, T. G. Pedersen, and B. K. Nikolić, arXiv:1512.06846.
[3] H. Mera. T. G. Pedersen, and B. K. Nikolić, Phys. Rev. Lett. 115, 143001 (2015).
Contributed talk
Some aspects of electron-phonon interactions in molecular junctions: Toward a description of time-dependent quantum transport
Laboratoire Ondes et Matière d'Aquitaine (LOMA), Université de Bordeaux et CNRS, 351 Cours de la Libération, 33405 Talence, FRANCE
Molecular electronics represents the ultimate limit of the "up-bottom" miniaturization process initiated by the semiconductor industry. Along this path, the laws of quantum mechanics have been found to impose fundamental limitations, especially in the ultimate case of molecular and atomic sized electronic junctions.
Despite several decades of investigations, a global understanding of current-voltage characteristics I(V), mechanical stability and heat losses of such molecular junctions is still lacking. A key mechanism for understanding all this phenomena lies in the coupling between electronic degrees of freedom (giving rise to an electronic current) and local vibrational modes of the junction (phonons giving rise to dissipation).
This presentation aims at reviewing recent advances in this field of research. In particular, we will show that, due to inelastic scattering with phonons, some information about electronic and vibrational properties of the junction, electronic correlations and effective lattice temperature is encoded into the electrical current noise curves ("shot-noise"), that is not present in standard I(V) characteristics. This opens up new technological perspectives and complementary spectroscopic tools at the nanoscale. We will present the first experimental evidence of this phenomenon [1] as well as its theoretical analysis, based on recent first-principle calculations [2]. Finally, we will provide a general picture and explore new directions and open questions arising from recent theoretical advances [3] dealing with the investigation of time-dependent quantum transport using NEGF techniques in the (ultra-fast) transient regime.
[1] M. Kumar, R. Avriller, A. Levy Yeyati and J. M. van Ruitenbeek, Phys. Rev. Lett. 108, 146602 (2012).
[2] R. Avriller and T. Frederiksen, Phys. Rev. B 86, 155411 (2012).
[3] R. Seoane Souto, R. Avriller, R. C. Monreal, A. Martin-Rodero and A. Levy Yeyati, Phys. Rev. B 92, 125435 (2015).
Contributed talk
Improved auxiliary-mode approach to time-dependent electron transport
Max Planck Institute for the Physics of Complex Systems, Nöthnitzer Str. 38, Dresden, Germany
The theoretical description of time-resolved phenomena in nanoscale devices remains a very challenging task. Several formalisms are available, but efficient approaches for general-purpose computations are still under investigation [1]. In Ref. [2] it was proposed to use an auxiliary-mode expansion of the self-energies arising in the time-dependent non-equilibrium Green's functions (TD-NEGF) formalism. To this end the Fermi function and the level-width function are expanded in terms of simple poles in the complex plane. By means of this parametrization, the equations of the NEGF theory, in particular the integro-differential equation for the reduced density-matrix, are converted into a set of coupled differential equations for auxiliary matrices.
This method is suited to describe time-dependent effects which stem from coupling the electronic system to other degrees of freedom, e.g., in the case of molecular systems where the influence of vibrational modes on electron transport plays a prominent role. Moreover, this approach has been used to study, e.g., electron transport through long molecular chains in the presence of thermal fluctuations from surrounding solvent molecules [3], or to characterize dynamical interference effects through meta- and para-aligned benzene molecules [4].
In the present contribution we discuss an improved new version of the auxiliary-mode approach, where the auxiliary matrices are replaced by vectors or scalars. This drastically reduces the overall computational effort and memory requirement. We demonstrate the method using a molecular wire as well as a closed ring structure, where we discuss propagation and interference of Leviton wave-packets.
[1] See for example: A.-P. Jauho, N. S. Wingreen and Y. Meir, Phys. Rev. B 50, 5528 (1994). S. Kurth et al., Phys. Rev. B 72, 035308 (2005). For a recent overview of the field see: B. Gaury et al., Phys. Rep. 534, 1 (2014) and references therein.
[2] A. Croy and U. Saalmann, Phys. Rev. B 80, 245311 (2009).
[3] B. Popescu et al., Phys. Rev. Lett. 109, 176 802 (2012).
[4] S. G. Chen et al., Phys. Chem. Lett. 5, 2748 (2014).