Click on the author name to see the abstract.
| 08:00-09:00 | Registration |
| Chair: Massimiliano Di Ventra | |
| 09:00-09:35 | Ferrer |
| 09:35-10:00 | Fediai |
| 10:00-10:25 | Palacios |
| 10:25-11:00 | Coffee break |
| Chair: Uri Peskin | |
| 11:00-11:35 | Stokbro |
| 11:35-12:10 | Pecchia |
| 12:10-12:35 | Novotny |
| 12:35-13:00 | Dridi |
| 13:00-15:30 | Lunch break |
| Chair: Vincent Meunier | |
| 15:30-16:05 | Papior |
| 16:05-16:30 | Brandimarte |
| 16:30-17:00 | Coffee break |
| Chair: Dvira Segal | |
| 17:00-17:35 | Di Ventra |
| 17:35-18:10 | Lambert |
| 18:10-18:35 | Nozaki |
Invited talk
GOLLUM: an efficient tool for ab-initio molecular electronics with non-trivial examples
1Faculta de Ciencias, Univerdad de Oviedo, 33007 Oviedo, Spain
2Research Centre on Nanomaterials and Nanotechnology - CINN
I will present in the first part of this talk the transport code GOLLUM, describing some of its non-trivial functionalities with specific examples. The second part of the talk will be used to describe simulations of the self-assembly of iso-cyano functionalized molecules in the presence of gold electrodes.
Contributed talk
New combination of DFT and Green function formalism pushes the limits of extended contacts simulations
1Institute for Materials Science and Max Bergmann Center of Biomaterials, TU Dresden, Germany
2Center for Advancing Electronics Dresden, TU Dresden, Germany
3Dresden Center for Computational Materials Science, TU Dresden, 01062 Dresden, Germany
The so-called extended contacts between metallic electrodes and materials with the reduced dimensionality are practically and theoretically important systems. Common examples are side-contacts between the carbon nanotube and metallic electrodes in carbon nanotube field-effect transistors (CNTFETs), and planar contacts between the graphene and metallic electrodes in graphene field-effect transistors (GFETs). A very active field of research are novel 2D materials that can be used as a channel of field-effect transistors (metal dichalcogenides, black phosphorus, etc.), and they need a proper theoretical treatment. What is common for all mentioned contacts is that the electric current can enter the corresponding 2D material or the tube gradually at a distance of up to 100 nm, which has been proven experimentally for CNTFETs and GFETs [1, 2].
Such a distributed current injection in extended contacts differs strongly from that occurring in the common set-ups considering in molecular electronics, which imply that the central part of the device has a local contact to the leads. If we would treat the device with extended contacts within the conventional combination of the DFT and NEGF formalism, we would need to simulate explicitly a 100 nm long system, which is numerically prohibitively expensive.
However, we have shown how to overcome this seemingly unresolvable issue in [3], and have applied this approach in order to predict the scaling of the contact resistance in the CNTFETs [4]. Here we show an application of the approach [3] to graphene-metal contact in GFETs. Figure 1 (a, b) shows geometry of the simulated system, which is a graphene sheet of top of two TiN electrodes. Figure 1(c) shows the scaling of the contact resistivity for three different metals (Pd, Ni, Ti) and one alloy (TiN) calculated from the first principles.
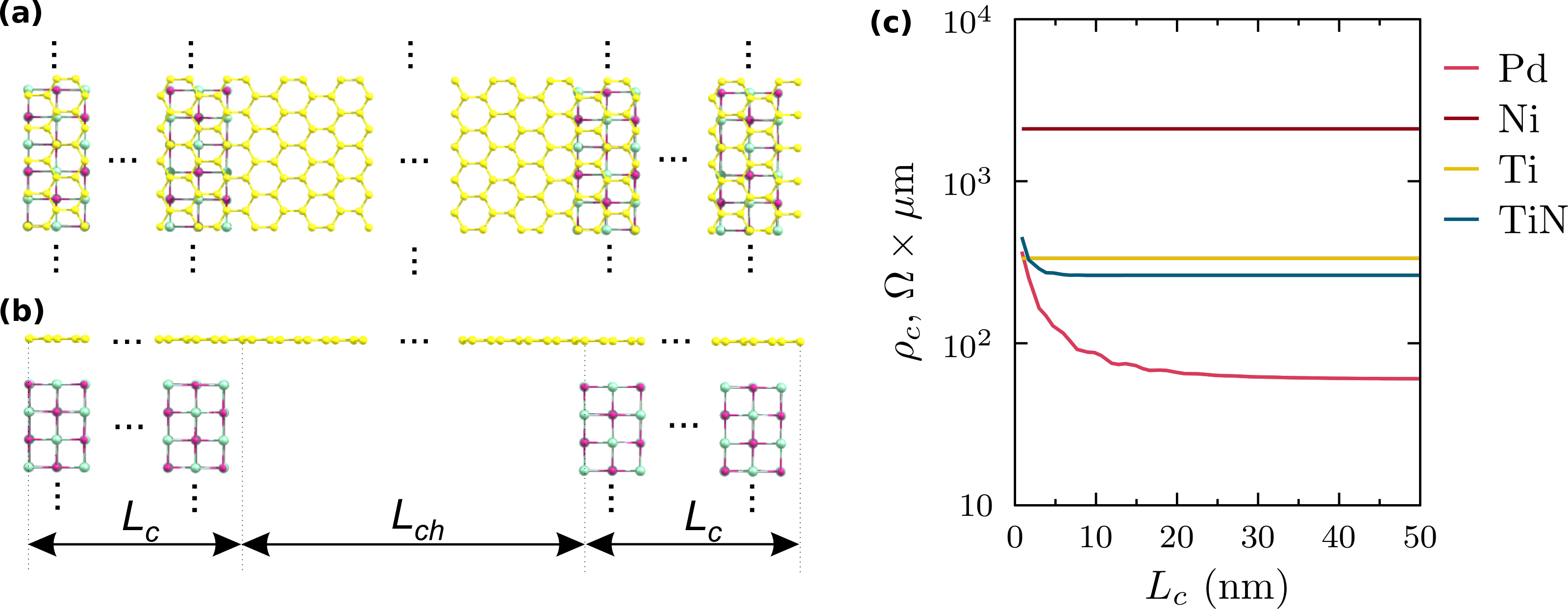
Figure 1: Top (a) and front (b) view of the GFET based on extended graphene – TiN contact. Simulated dependence of the contact resistivity of the GFET as a function of contact length for three metal and one alloy (c).
[1] A. Franklin et al., ACS Nano 8 (7), 7333 (2014).
[2] A. Franklin et al., IEEE Electron Dev. Lett. 32 (8) (2011).
[3] A. Fediai et al., Phys. Rev. B. 91, 165404 (2015).
[4] A. Fediai et al., (accepted manuscript) Nanoscale (2016).
Contributed talk
A fully first-principles molecular orbital approach to the Kondo effect in organic molecules
1Departamento de Física de la Materia Condensada and Condensed Matter Physics Institute (IFIMAC), Universidad Autónoma de Madrid Campus de Cantoblanco, 28049 Madrid, Spain
2Institute of Condensed Matter and Nanosciences (IMCN) Université Catholique de Louvain (UCL) Chemin des étoiles 8, B-1348 Louvain-la-Neuve, Belgium
3Max-Planck-Institut für Mikrostrukturphysik Weinberg 2, 06120 Halle, Germany
We present a generic parameter-free approach to the Kondo problem, which is particularly suitable for carbon-based molecules or organic radicals in contact to metal electrodes. In these molecules the screened spin may originate in an unpaired delocalized π electron. Our proposed method is structured in three distinct stages. In the first stage we perform a density functional theory (DFT) calculation for an open system consisting of the molecule attached to infinite electrodes. Second, we make use of the DFT results to obtain the three fundamental ingredients of an Anderson impurity model (AIM), i.e., the energy of the impurity levels (molecular orbitals here), the hybridization function, and the electron--electron interactions. In the final stage we solve the AIM through an impurity solver to include the dynamical correlations. This parameter-free theory is successfully tested on the case of a C60 molecule in a transistor configuration (see Fig.1), correctly reproducing the zero-bias features arising in the charge sector 1 (spin-1/2 Kondo), and in the charge sector 2 where a singlet-triplet quantum phase transition has been reported experimentally (see Fig. 2 and Ref. 1).
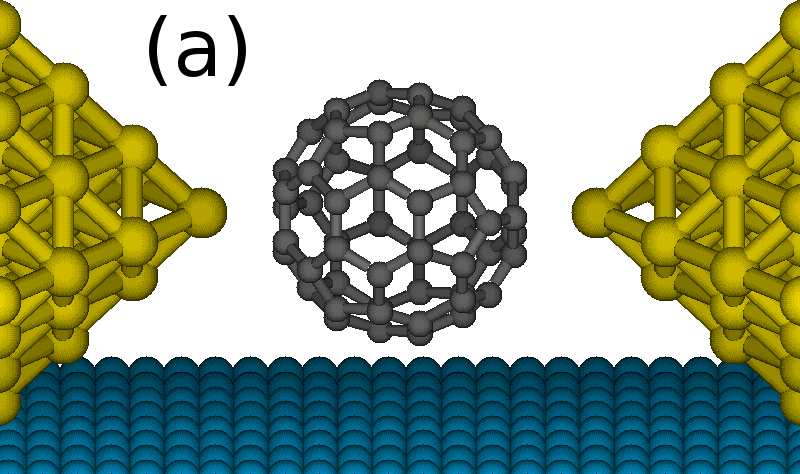
Figure 1. C60 molecule in a transistor configuration
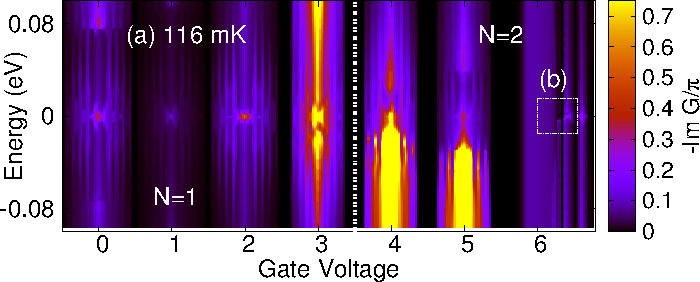
Figure 2. Calculated spectral function as function of gate voltage.
[1] N. Roch, S. Florens, V. Bouchiat, W. Wernsdorfer, and F. Balestro, Nature 453, 633 (2008).
Invited talk
Simulation of single electron molecular transistors
1QuantumWise A/S, Fruebjergvej 3, 2100 Copenhagen, Denmark
2Copenhagen University, Denmark
In this presentation we present simulations with the Atomistix ToolKit of a molecule weakly coupled with metallic electrodes [2]. The device is operating in the incoherent regime, i.e. a single electron molecular transistor. The theoretical methodology of the simulation is illustrated for a benzene and C60 molecular transistor [2]. The main objective is the simulation of a single OPV5-tBu molecule between gold electrodes. This system was previously experimentally investigated by Kubatkin et al. [1]. In order to directly simulate the experimental geometry we have developed a finite-element DFT method that allows efficient treatment of complex molecules interacting with arbitrarily large electrostatic environments [3]. The details of the implementation will be discussed. The simulations are compared with experimental data of Kubatkin et al. [1]. We find quite different properties than in the experiment and point to a number of possible reasons for the discrepancy.
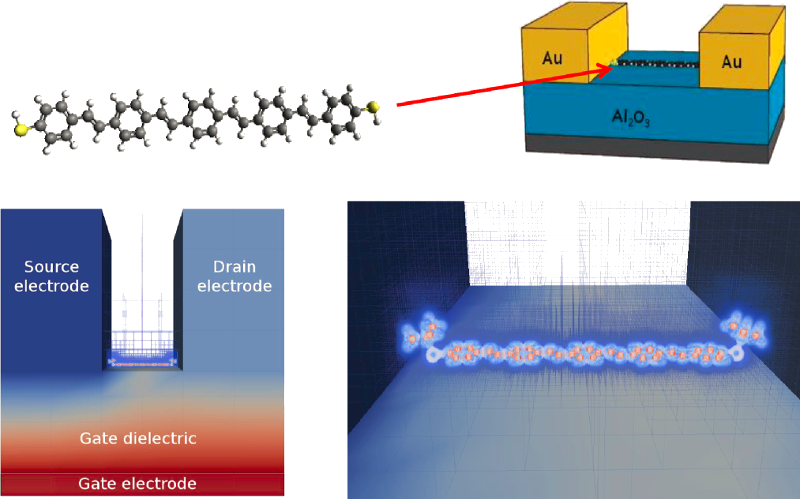
Figure 1: (top) OPV5 transistor setup. (bottom) The simulation setup with the finite element mesh. The mesh is fine around the molecule and coarse away from the molecule.
[1] S. Kubatkin et al., Nature 425, 698 (2003).
[2] K. Stokbro, Journal of Physical Chemistry C 114, 20461–20465 (2010).
[3] J. Avery, PhD Thesis, Copenhagen University (2011).
Invited talk
Transport in molecular junctions beyond the coherent picture
1Consiglio Nazionale delle Ricerche, ISMN, Via Salaria Km 29.6, 00017 Monterotondo, Rome, Italy
2Nano-Science Center and Department of Chemistry, University of Copenhagen, Universitetsparken 5, 2100 Copenhagen Ø, Denmark
3BCCMS, Bremen Centre of Computational Materials Sciance, Univ. Bremen, Germany
4Department of Mechanical Engineering, Massachusetts Institute of Technology, 77 Massachusetts Avenue, Cambridge, Massachusetts 02139, USA
5Technische Universität München, Electrical Engineering and Information Technology, Arcisstr. 21, 80333 München, Germany
Molecular electronics has opened many field of research and it is still a rather lively subject of investigations across Europe. In this talk coherent and incoherent transport in molecular junctions based on the non-equilibrium Green's function formalism and the density-functional tight-binding method will be discussed [1]. Transport in controlled STM junctions can lead to unexpected transport behaviors due to conformational changes of the molecule induced by the tip. The interplay between interference and electron-vibron scattering in cross-conjugated molecules will be presented [2-4].
[1] A. Pecchia, G. Penazzi, L. Salvucci, and A. Di Carlo, New Journal of Physics 10, 065022 (2008).
[2] J. Lykkebo, A. Gagliardi, A. Pecchia, and G. C. Solomon, ACS Nano 7, 9183-9194 (2013).
[3] J. Lykkebo, A. Gagliardi, A. Pecchia, and G. C. Solomon, J. Chem. Phys. 141, 124119 (2014).
[4] J. Lykkebo, G. Romano, A. Gagliardi, A. Pecchia, and G. C. Solomon, J. Chem. Phys. 144 (2015).
Contributed talk
Quantum interference effects on IETS signals
1Department of Condensed Matter Physics, Faculty of Mathematics and Physics, Charles University in Prague, Czech Republic
2Department of Physics, University of Konstanz, Germany
We study characteristics of the IETS signal in molecules exhibiting destructive quantum interference in the elastic transmission such as meta-benzene or cross-conjugated molecules. We formulate a simple propensity rule based on the Hückel model for the molecule, which states that antisymmetric vibrational modes do not contribute to the IETS spectra. This analytical rule is verified by full ab-initio calculations based on DFT [1,2]. Our results are at variance with previous calculations by Lykkebo et al. [3,4] claiming strong suppression of all fundamental vibrational modes. In fact, we observe that for closely related pairs of molecules with/without the destructive interference features (e.g., meta- vs. para-benzene or linearly vs. cross-conjugated) the IETS spectra are very similar (apart from an overall decrease of magnitude by a factor ~10 for molecules with interference features), see Fig. 1, or even those for molecules with interference features are richer (case of benzene, not shown). We trace this discrepancy in predictions to the differences in the numerical evaluation of the electron-vibration coupling elements by various IETS codes [2,5]. These findings raise the issue of the reliability and reality of calculations of electron-vibration interaction.
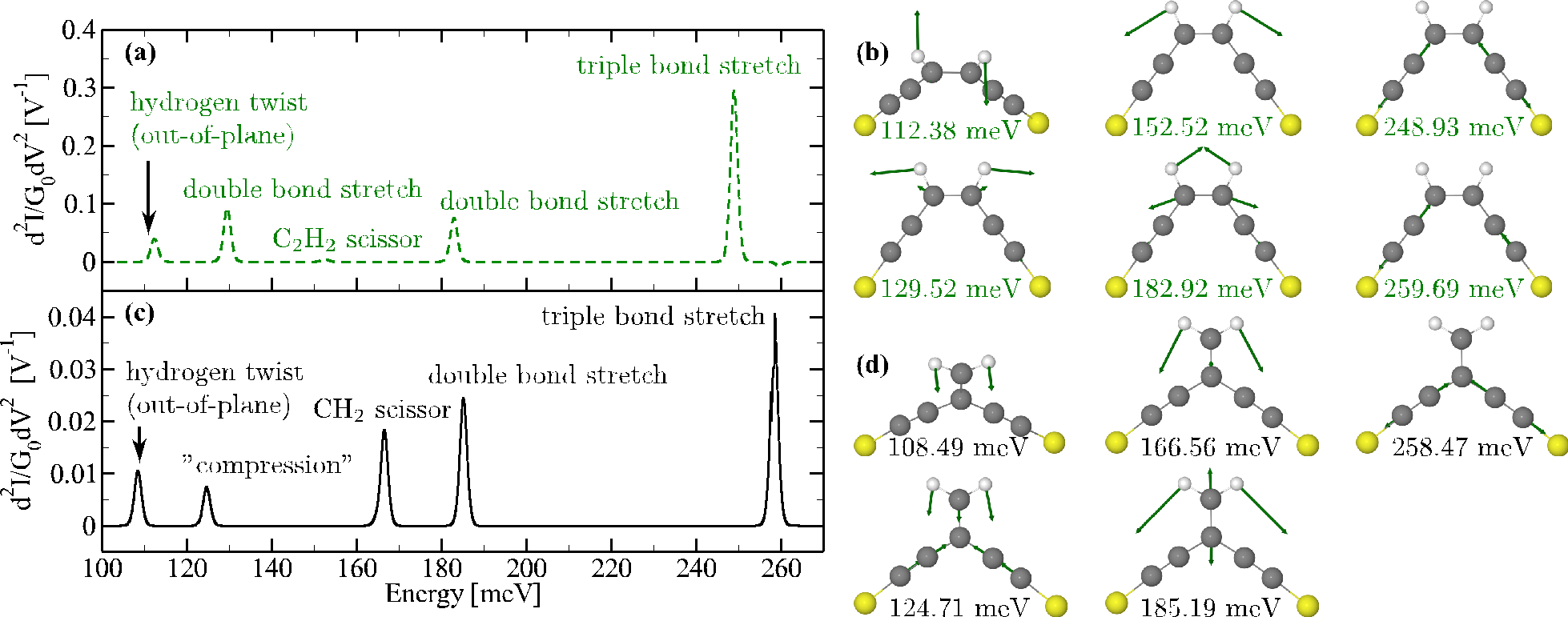
Figure 1: IETS signals and corresponding vibrational modes for a linearly conjugated molecule (a,b) with no destructive interference and for a cross-conjugated molecule (c,d) exhibiting the destructive interference in the elastic transmission. Apart from an overall scaling down of the magnitude the two sets show no dramatic changes in the character.
[1] J. K. Viljas et al., Phys. Rev. B 72, 245415 (2005).
[2] M. Bürkle et al., Phys. Status Solidi B 250, 2468 (2013).
[3] J. Lykkebo et al., ACS Nano 7, 9183 (2013).
[4] J. Lykkebo et al., J. Chem. Phys. 141, 124119 (2014).
[5] A. Gagliardi et al., Phys. Rev. B 75, 174306 (2007).
Contributed talk
A novel design of quantum half adder using Quantum Hamiltonian Computing (QHC) approach: comparative study with the Qubit half adder
CEMES-CNRS, 29 Rue J. Marvig, 31055 Toulouse Cedex, France
Nowadays, the realization of reversible arithmetic and logical operation using quantum mechanical phenomenon is one of the most challenging problems in modern technology. However, it is commonly believed that a large number of quantum states is needed to realize even small quantum logic gates. This large number of quantum states provides a major obstacle for building more complex logic functions and serious barrier for a full-scale implementation because the larger the quantum system is, the greater the interaction with the external environment will be important causing the loss of information. Thus a superior approach is needed to minimize the total number of states required to realize these quantum logic gates. The Quantum Hamiltonian Computing (QHC) approach appeared to be a good candidate to construct complex quantum circuits with a minimum number of states [1]. The capability of QHC approach to minimize the total number of quantum states comes from the fact that we do not need that the quantum system be divided in qubits. Therefore, we can take the advantage of the total quantum system to perform a logic function but with a minimum spatial extension of the quantum system.
In this presentation, we compare the QHC approach with the Qubit approch through the study of the standard quantum half adder. We will show first of all how the QHC approch is able to greatly minimize the number of states required to construct this complex logic funcion. We willl show in particular that only a 4 states are needed to construct a QHC half adder. We also show that the measurement of the frequency [2] instead of the population provides another advantage agains Qubit in terms of robustness of the logic gates. we will discuss for each approch the energy efficiency of the gate as the minimum energy needed to perform the calculation and the speed of calculation.
[1] G. Dridi, R. Julien, M. Hliwa, and C. Joachim, The mathematic of a Quantum Hamiltonian Compulsion half adder Boolean logic gates, Nanotechnologie 34, 344003 (2015).
[2] N. Renaud and C. Joachim, Classical Boolean logic gates with quantum systems, J. Phys. A: Math and Theor. 44, 15 (2011).
Invited talk
N≥1 NEGF calculations & molecular projected transport
1Catalan Institute of Nanoscience Nanotechnology, Campus UAB, Bellaterra, Spain
2Center for Nanostructured Graphene (CNG), DTU Nanotech, DTU, Denmark
I will present the recent development of N≥1 electrode NEGF simulations which enables simulations of multiprobe physics. The specific implementation will be presented [1,2] and the efficiency and scalability of the code (TranSIESTA) will be emphasized. The code implements hybrid parallelization which easily enables efficient calculations of systems with more than 10,000 orbitals under non-equilibrium, see Fig. 1 which shows the maximum memory usage with respect to system size (x-axis) and electrode size (y-axis), left. The right shows the computation time of a single SCF with NEGF. Using the N electrode capability I will present initial 3 terminal results based on a 2D-graphene interconnects [3].
After having introduced the NEGF method I expand on the transmission calculations where I will focus on projected molecular transport, see Fig. 2. Projections enable spectroscopy of molecular eigenstates and the identification of the electron carrying pathways. They will provide an intuitive understanding of the physics governing the transport properties of the junction. I will analyze the transport projection of the C60 molecule in a close packed Cu surface [4]. Importantly the projection is not limited to molecules and it will be shown that a generic projection onto k-resolved eigenstates is also possible.
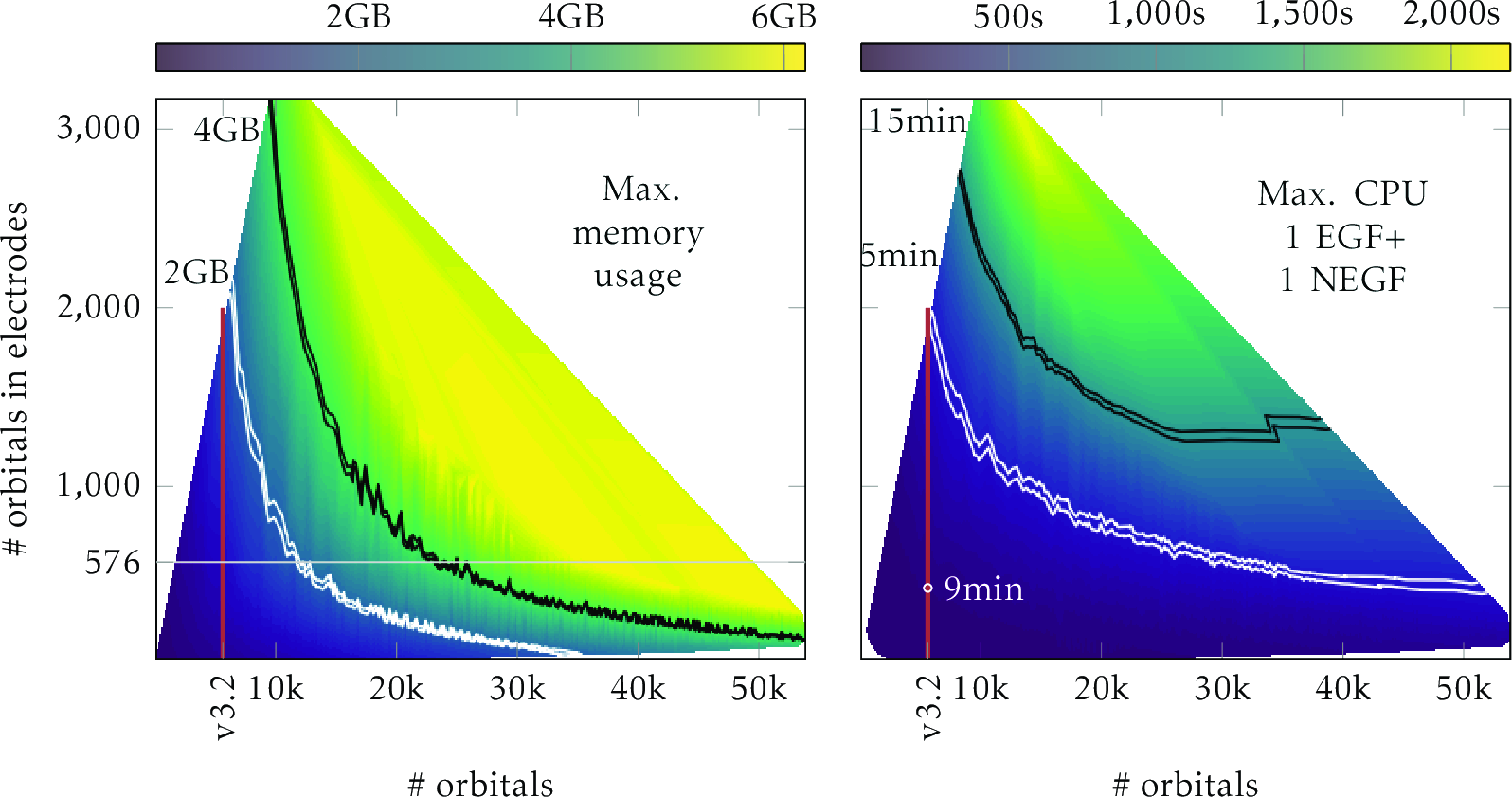
Fig. 1: Memory/Time use of 2-terminal NEGF calculations.
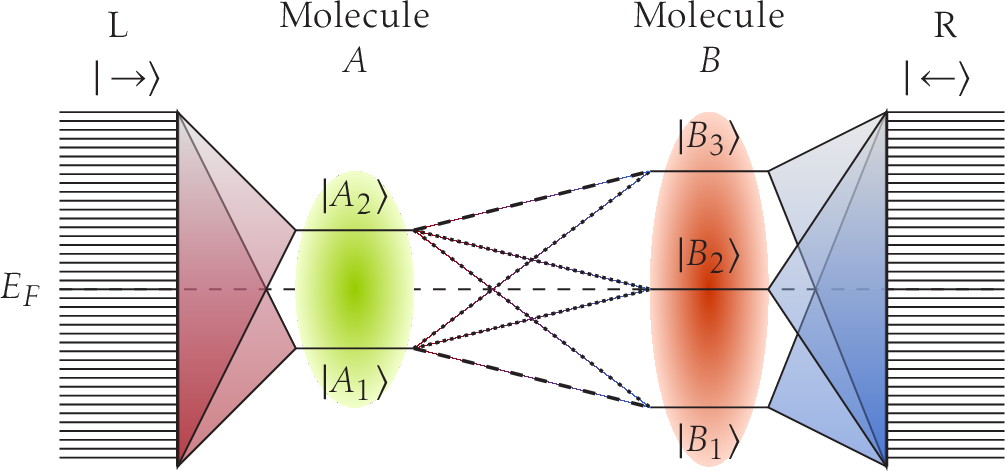
Fig. 2: Transmission projection of molecule eigenstates.
[1] Papior, PhD thesis (2016).
[2] Papior et al. (to be submitted).
[3] Jacobsen et al., Carbon 101, 101-106 (2016).
[4] Schneider et al., Journal of Physics Condensed Matter 27, 015001 (2014).
Contributed talk
Aspects of graphene nanoribbon device simulations
1Centro de Física de Materiales (CFM), Spain
2Donostia International Physics Center (DIPC), Spain
3Catalan Institute of Nanoscience Nanotechnology - Campus UAB, Spain
4Center for Nanostructured Graphene (CNG) - DTU Nanotech, Denmark
5IKERBASQUE, Basque Foundation for Science, Spain
6CIC nanoGUNE, Spain
7Department of Physics - University of Basel, Switzerland
8PRESTO - Japan Science and Technology Agency, Japan
9Department of Chemistry from Graduate School of Science - Nagoya University, Japan
10Institute of Transformative Bio-molecules - Nagoya University, Japan
11CREST - Japan Science and Technology Agency, Japan
Graphene nanoribbons (GNRs) have been considered as strong candidates for electronics, since they incorporate some of the remarkable properties from graphene while presenting a band gap [1]. These structures can be fabricated nowadays with a high control on the edge structure with the so-called bottom-up approaches [2]. Together with these experimental breakthrough a major effort has been devoted to the theoretical and computational methods developments. In this talk I will show some challenging features found when simulating GNRs based devices, such as: 1. The effects on the transport properties of 4-terminal crossed GNRs devices when varying the structural parameters determining the crossing; 2. The effect of substitutional chemical defects on the transmission channels and the importance of a proper description of the electrostatics in quasi 1-dimension systems.
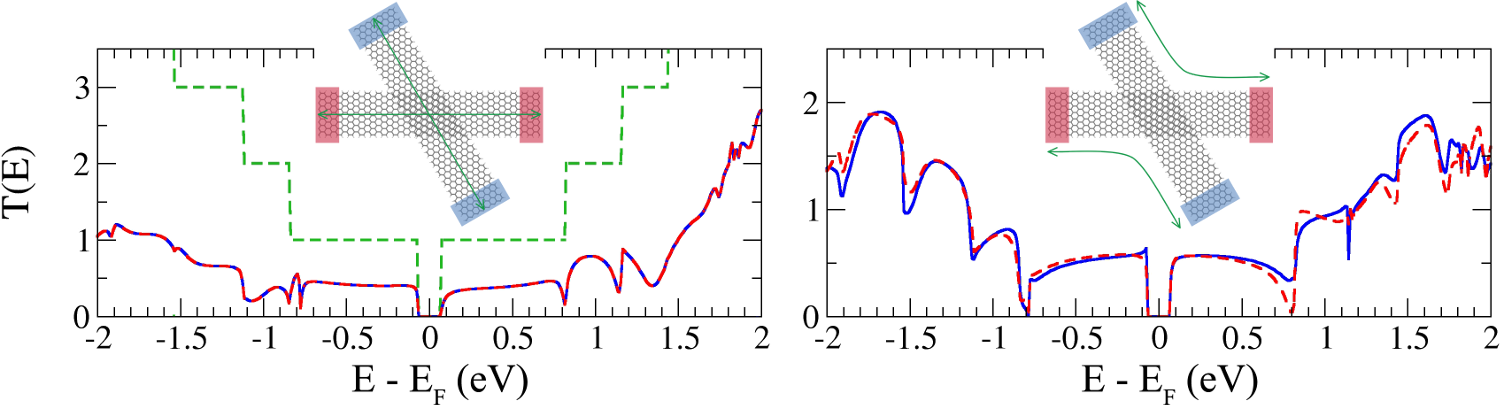
Figure 1: Direct (left) and inter-ribbon (right) transmissions at V=0 in a crossbar system with 60˚ rotation angle.
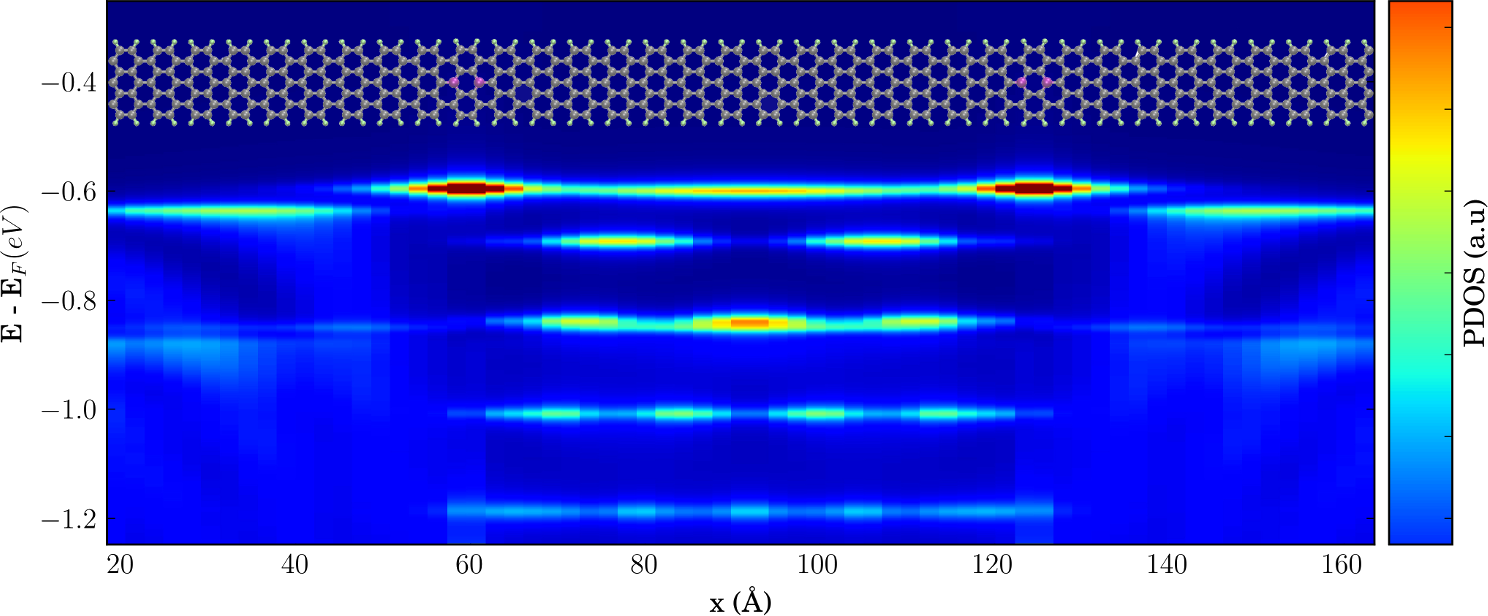
Figure 2: Project density of states from a substitutional boron-doped armchair graphene nanoribbon.
[1] L. Yang et al., Phys. Rev. Lett. 99, 186801 (2007).
[2] J. Cai et al., Nature 466, 470-473 (2010).
Invited talk
Functional theories of thermoelectric phenomena
Department of Physics, University of California San Diego, La Jolla, CA 92093-0319 USA
I will discuss non-equilibrium density functional theories of local temperatures and associated heat currents that are particularly suited for the study of thermoelectric phenomena. In one case, I will introduce a functional theory of open quantum systems [1] that allows for the study of local temperatures by the introduction of local thermal probes. In another [2], we couple the local temperature field to an energy density operator. I will also provide predictions on the local temperature oscillations in atomic wires [3], carbon nano-ribbons and graphene junctions [4], and discuss similarities and differences between the different local temperature definitions in the strongly-correlated regime [5].
[1] M. Di Ventra and R. D'Agosta, Phys. Rev. Lett. 98, 226403 (2007).
[2] F. Eich, G. Vignale and M. Di Ventra, Phys. Rev. Lett. 112, 196401 (2014).
[3] Y. Dubi and M. Di Ventra, Nano Lett. 9, 97 (2008).
[4] J. P. Bergfield, M. A. Ratner, C. A. Stafford, and M. Di Ventra, Phys. Rev. B 91, 125407 (2015).
[5] L. Z. Ye, D. Hou, X. Zheng, Y.J. Yan, and M. Di Ventra, Phys. Rev. B 91, 205106 (2015).
Invited talk
Quantum-interference-enhanced thermoelectricity in single molecule junctions
Department of Physics, Lancaster University, UK
Although the dream of manipulating quantum interference in single molecules has been discussed for many years, experimental evidence of the effect of quantum interference on the room-temperature electrical conductance of single-molecules was reported only recently [1].
In this talk, I will present a brief outline of recent advances in our understanding of quantum interference in single-molecules [2] and then discuss how quantum interference can be exploited to increase the thermoelectric performance of single molecules [3] and assemblies of molecules connected to nano-gap electrodes.
[1] J. Am. Chem. Soc. 133, 11426, (2011); Nature Nano. 7, 305 (2012); Nat. Nano. 7, 663 (2012); Phys. Rev. Lett. 109, 056801 (2012); Nano. Lett. 6, 1643-1647 (2012); J. Am. Chem. Soc. 134, 5262 (2012); Beilstein J. Nanotech. 2, 699 (2011) and refs. therein.
[2] C. J. Lambert, Chem. Soc. Rev. 44, 875-888 (2015); Y. Geng et al., J. Am. Chem. Soc. 137, 4469 (2015); Sangtarash et al., J. Am. Chem. Soc. 137, 11425 (2015); D. Manrique, et al., Nature Comm. 6, 6389 (2015); Berritta et al, Nanoscale 7, 1096 (2015).
[3] Evangeli et al., Nano Lett. 13, 2141-2145 (2013); Garcia-Suarez et al., Nanotechnology 25, 205402 (2014); Rincón-García et al., Nature Mat. 15, 289 (2016); Sadeghi et al., Nano Lett. October (2015) DOI: 10.1021/acs.nanolett.5b03033; Ismael et al., Nanoscale 7 17338 (2015).
Contributed talk
Quantum interference in open quantum systems: design of transmission spectra and analysis on thermoelectric properties
1Department of Theoretical Physics, University of Paderborn, Germany
2Institute for Materials Science, TU Dresden, Germany
Quantum interference (QI) in molecular devices has drawn an increasing attention in the last decades because of unique features observed in conductance spectra applicable to thermoelectric devices and molecular switches [1-3]. In order to tailor the molecular devices exploiting QI effects, it is of great theoretical and practical interest to clarify the relationship between the line shape of conductance spectra including QI features and controlling parameters such as electronic structures (on-site energies and transfer integrals) and topologies of molecules.
In this talk, we present a simple graphical method (parabolic diagram) that we developed to visualize the relation between the electronic parameters and line shape of conductance spectrum influenced by QI effect in the molecular junction with a side group (T-shaped molecule, Fig. 1) [4,5]. We will also present systematic analysis of thermoelectric properties in a series of molecular junctions having different topologies and QI features in toy model basis [6]. In this work, we consider a homogeneous toy model where all on-site energies are identical and model four types of molecular junctions due to their topological connectivities. We systematically analyze their transmission spectra, density of states, and thermoelectric properties. Even without the degree of freedom of on-site energies an asymmetric Fano peak could be realized in the homogeneous systems with the cyclic configuration. We also calculate the thermoelectric properties of the model systems with and without fluctuation of on-site energies. Even under the fluctuation of the on-site energies, the finite thermoelectrics are preserved for the Fano resonance, thus cyclic configuration is promising for thermoelectric applications.
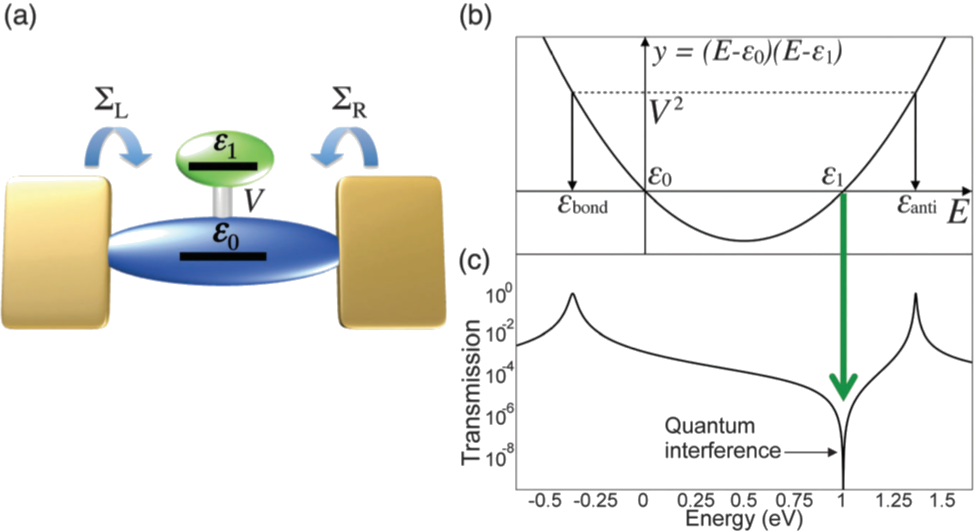
Figure 1: (a) Electronic parameters in a T-shaped molecule. (b) A parabolic diagram. Drawing a parabola with set of parameters (ε0, ε1 and V) allows one to visualize the relationship between (anti-) resonances and the electronic parameters as shown in panel (c).
[1] S. V. Aradhya and L. Venkataraman, Nat. Nanotechnol. 8, 399 (2013).
[2] W. Hong et al., Beilstein J. Nanotechnol. 2, 699 (2011).
[3] C. M. Guédon et al. Nat. Nanotechnol. 7, 305 (2012).
[4] DN, H. Sevinçli, S. M. Avdoshenko, R. Gutierrez, G. Cuniberti, J. Phys. Conf. Ser. 427, 012013 (2013).
[5] DN, H. Sevinçli, S. M. Avdoshenko, R. Gutierrez, G. Cuniberti, PCCP 15, 13951 (2013).
[6] DN, S. M. Avdoshenko, H. Sevinçli, G. Cuniberti J. Appl. Phys. 117, 074308 (2014).