Click on the author name to see the abstract.
| Chair: Fernando Delgado | |
| 09:00-09:35 | Pauly |
| 09:35-10:00 | Baugh |
| 10:00-10:25 | Garcia-Lekue |
| 10:25-11:00 | Coffee break |
| Chair: Antti-Pekka Jauho | |
| 11:00-11:35 | Peskin |
| 11:35-12:10 | Wang |
| 12:10-12:35 | Alberga |
| 12:35-13:00 | Costuas |
| 13:00-15:30 | Lunch break |
| Chair: Stefan Roche | |
| 15:30-16:05 | Thoss |
| 16:05-16:30 | Cucinotta |
| 16:30-18:30 | Poster session (soft drinks and coffee) |
Invited talk
Charge and energy transport in atomic and molecular junctions
Department of Physics, University of Konstanz, D-78457 Konstanz, Germany
Single-atom and single-molecule junctions represent the ultimate limit to the miniaturization of electrical circuits. They are also ideal platforms for testing quantum transport theories that are required to describe charge and energy transfer in novel functional nanometer-scale devices.
In this seminar, I will review the recent theoretical progress of my group towards the description of elastic and inelastic charge transport in atomic and molecular junctions as well as thermoelectric phenomena and heat dissipation. After a brief introduction, I will discuss results on the conductance [1,2], inelastic electron tunneling spectra [3,4] and the thermopower [5-8]. Next, I will show an ab-initio characterization of the thermoelectric figure of merit of molecular junctions including both electronic and phononic contributions to the thermal conductance [9]. Finally, I will present combined experimental and theoretical efforts to understand the heat dissipation in atomic-scale junctions [10,11].
[1] Influence of conformation on conductance of biphenyl-dithiol single-molecule contacts, A. Mishchenko, D. Vonlanthen, V. Meded, M. Bürkle, C. Li, I. V. Pobelov, A. Bagrets, J. K. Viljas, F. Pauly, F. Evers, M. Mayor, and T. Wandlowski, Nano Lett. 10, 156 (2010).
[2] Conduction mechanisms in biphenyl-dithiol single-molecule junctions, M. Bürkle, J. K. Viljas, A. Mishchenko, D. Vonlanthen, G. Schön, M. Mayor, T. Wandlowski, and F. Pauly, Phys. Rev. B 85, 075417 (2012).
[3] Influence of vibrations on electron transport through nanoscale contacts, M. Bürkle, J. K. Viljas, T. J. Hellmuth, E. Scheer, F. Weigend, G. Schön, F. Pauly, Phys. Status Solidi B 250, 2468 (2013).
[4] Identification of the current path for a conductive molecular wire on a tripodal platform, M. A. Karimi, S. G. Bahoosh, M. Valášek, M. Bürkle, M. Mayor, F. Pauly, and E. Scheer (submitted).
[5] Ab initio study of the thermopower of biphenyl-based single-molecule junctions, M. Bürkle, L. A. Zotti, J. K. Viljas, D. Vonlanthen, A. Mishchenko, T. Wandlowski, M. Mayor, G. Schön, F. Pauly, Phys. Rev. B 86, 115304 (2012).
[6] Length-dependent conductance and thermopower in single-molecule junctions of dithiolated oligophenylene derivatives, F. Pauly, J. K. Viljas, and J. C. Cuevas, Phys. Rev. B 78, 035315 (2008).
[7] Molecular dynamics study of the thermopower of Ag, Au, and Pt nanocontacts, F. Pauly, J. K. Viljas, M. Bürkle, M. Dreher, P. Nielaba, J. C. Cuevas, Phys. Rev. B 84, 195420 (2011).
[8] Quantum thermopower of metallic atomic-size contacts at room temperature, C. Evangeli, M. Matt, L. Rincón-García, F. Pauly, P. Nielaba, G. Rubio-Bollinger, J. C. Cuevas, N. Agraït, Nano Lett. 15, 1006 (2015).
[9] First principles calculation of the thermoelectric figure of merit for [2,2]paracyclophane-based single-molecule junctions, M. Bürkle, T. J. Hellmuth, F. Pauly, Y. Asai, Phys. Rev. B 91, 165419 (2015).
[10] Heat dissipation in atomic-scale junctions, W. Lee, K. Kim, W. Jeong, L. A. Zotti, F. Pauly, J. C. Cuevas, P. Reddy, Nature 498, 209 (2013).
[11] Heat dissipation and its relation to thermopower in single-molecule junctions, L. A. Zotti, M. Bürkle, F. Pauly, W. Lee, K. Kim, W. Jeong, Y. Asai, P. Reddy, and J. C. Cuevas, New J. Phys. 16, 015004 (2014).
Contributed talk
Vibrationally-mediated single molecule switching studied by low-temperature STM
1Fritz-Haber Institute of the Max-Planck Society Faradayweg 4-6, 14195, Berlin, Germany
2Institute of Physical Chemistry, Polish Academy of Sciences, Kasprzaka 44/52, Warsaw 01-224, Poland
3Faculty of Mathematics and Natural Sciences, College of Science, Cardinal Stefan Wyszyński University, Dewajtis 5, 01-815 Warsaw, Poland
4Surface Science Research Centre and Department of Chemistry, University of Liverpool, Liverpool L69 3BX, UK
Recent experimental results for the intra-molecular hydrogen-transfer reaction, tautormization, of a single Porphycene molecule on the Cu(110) surface will be reported. The reversible cis-cis switching is directly observed and controlled by scanning tunneling microscopy (STM) and the reaction yield, probability per electron, is investigated as function of the tunneling current and bias voltage. The yield shows a steep increase at the energy of vibrational excitation of porphycene, and also exhibits a clear isotope effect when substituting the transferred H-atoms with deuterium, revealing a vibrationally mediated process. These results indicate that the reaction coordinate involves a skeletal motion of porphycene, particularly the "door-way" vibrational modes, that couples with the N-H(D) stretching mode and which shows a pronounced anharmonicity because of the intramolecular H-bonds. Therefore, the molecular swtitching process is an inherently multidimensional problem and a true understanding of the H-transfer resulting in the Cis-Cis transformation might require a multidimensional molecular dynamics calculation. Moreover, since the nuclear quantum effects like tunneling and zero-point energy would contribute to the process due to the small mass of H-atom, quantum mechanical treatments may also be required for the complete description of switching process. Specifically, a microscopic understanding electron-phonon coupling to induce vibrational excitation within a single molecule and how the dynamics of the subsequent IVR results in the excitation of the switching/reaction coordinate.
Contributed talk
Ab initio quantum transport calculations using plane waves
1Donostia International Physics Center (DIPC), Donostia-San Sebastián, Spain
2IKERBASQUE, Basque Foundation for Science, Bilbao, Spain
Since the advent of the scanning tunneling microscope (STM), a major effort has been devoted to the development of computational methods appropriate for the theoretical investigation of electron transport at the nanoscale. In particular, given the high sensitivity of conductance to atomic-structure details, density functional theory (DFT) has become an essential tool in transport calculations. The most commonly employed method combines the nonequilibrium Green's function (NEGF) with a DFT electronic structure, which are usually based on localized basis set. An alternative route is to explicitly solve the scattering states, although this approach is in most cases extremely expensive.
In this talk, I will describe a method to efficiently calculate the scattering states in quantum-transport problems using a plane-wave basis set [1]. Our approach is valid for any applied bias voltage since the current is calculated self-consistently, and can include exact evanescent states, which are also calculated using plane waves. The scattering states are solved exactly in a fast and numerically stable procedure with a computational time similar to a conventional ground state calculation. Based on these scattering states one can straightforwardly calculate the transmission coefficients and the corresponding electronic current.
Here, I will present several examples using this method, which range from electron tunneling to molecular electronics [2]. Our results demonstrate that our method is a good practical and accurate approach to study elastic quantum transport for a variety of problems. Particularly, it is useful to study tunneling through vacuum where the localized atomic orbital based method might face a difficulty to describe the electronic wave functions [3].
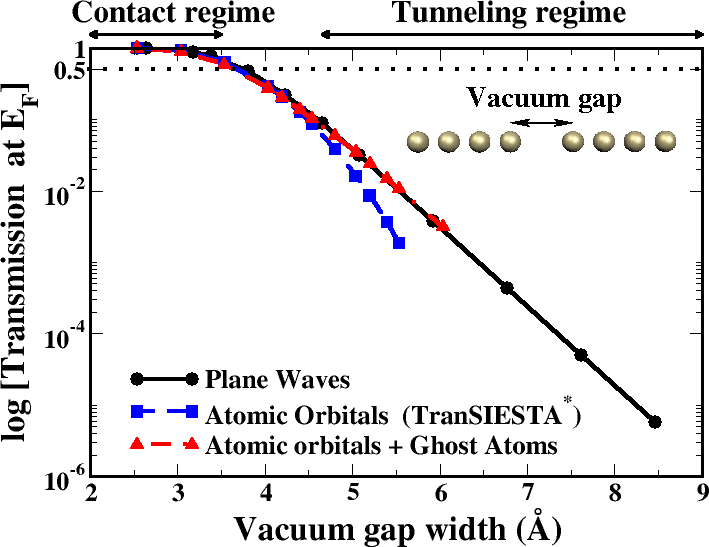
Figure: Total transmission at the Fermi level for a broken Au chain as a function of the vacuum-gap width obtained using plane waves (solid line), atomic orbitals (dashed line) and atomic orbitals with ghost atoms (dashed dotted line). [*] M. Brandbyge et al., Phys. Rev. B 65, 165401 (2002).
[1] A. Garcia-Lekue et al., Phys. Rev. B 74, 245404 (2006).
[2] A. Garcia-Lekue et al., Prog. in Surf. Sci. 90, 292 (2015).
[3] A. Garcia-Lekue et al., Phys. Rev. B 82, 035410 (2010).
Invited talk
Time-dependent phenomena in single molecule junctions: Coherent electron pumps, transient currents and the directional photo-electric effect
Schulich Faculty of Chemistry and the Lise Meitner Center for Computational Quantum Chemistry, Technion-Israel Institute of Technology, Haifa 32000, Israel
The study of charge and energy transport in single molecule junctions out of equilibrium introduces unique theoretical and experimental challenges. In the talk, theoretical studies of field-driven molecular junctions are presented which suggest new manifestations of unique single molecule properties in macroscopic observables. Having a molecule as the 'bottle neck' for charge and energy transport, the characteristic length and time scales suggest that coherent dynamics dominates the transport properties, and therefore the transport can be coherently controlled. In particular we shall focus on 'electron pumps' which convert radiation field into directed electronic currents. The principle of operation and the experimental manifestations will be analyzed theoretically under conditions ranging from a sudden pulse to cw excitation, and from direct excitation of the molecule to excitation through the leads plasmons. The latter suggest the existence of a single molecule-based directional photo-electric effect.
Invited talk
Nonequilibrium Green's function method and near-field radiation
Department of Physics, National University of Singapore
The nonequilibrium Green's function (NEGF) method is a powerful technique for studying quantum transport. We begin with a brief introduction of NEGF, emphasizing the role of contour order in defining the Green's functions. After a briefly mentioning of few applications, we discuss the problem of near-field radiation between metal plates. In dealing with the quantum field of the electromagnetism, an operator normal order is essential in using the Poynting vector to compute the radiation energy flux. We then present results of our model calculations.
[1] J.-S. Wang, J. Wang, and J. T. Lü, Quantum thermal transport in nanostructures, Eur. Phys. J. B 62, 381 (2008).
[2] J.-S. Wang, B. K. Agarwalla, H. Li, and J. Thingna, Nonequilibrium Green's function method for quantum thermal transport, Front. Phys. 9, 673 (2014).
Contributed talk
Hole transporter materials for organic photovoltaics: a theoretical approach
1Institut de Recherche de Chimie Paris CNRS Chimie ParisTech, PSL Research University, 11 rue P. et M. Curie, F-75005 Paris 05, France
2Dipartimento di Farmacia - Scienze del Farmaco, Università di Bari "Aldo Moro", Via Orabona, 4, I-70126 Bari, Italy
3INFN - Sez. di Bari and Dipartimento di Medicina Clinica e Sperimentale, University of Foggia, Viale Pinto, 71122 Foggia, Italy
Organic solar cells (OSC) based on π-conjugated polymers and organic molecules have seen increasing interest in recent years as an alternative to inorganic photovoltaics due to their possible low cost application in large area and flexible devices. The hole-transporter material (HTM) is one of the key components of an OSC having the function to transport the holes generated from the exciton dissociation to the electrical contact. In this context computational techniques can provide valuable insights in the characterization of electronic, optical and charge transport properties of a HTM. First we explore the relation between the morphological and the hole transport properties of P3HT and PBTTT polymers in crystalline phase [1]. Using molecular dynamics (MD) to simulate bulk supercells and the Marcus theory to calculate the hole mobility we show that PBTTT displays a larger charge mobility than P3HT due to the interdigitation of the side chains enhancing the stability of the conjugated rings on the backbones. Then we present combined MD-DFT calculations to investigate six triphenylamine-based hole-transporter materials [2] used in solid-state dye-sensitized solar cells (ssDSSC), including the state-of-the-art material in this field, spiro-OMeTAD. We find that all of the studied materials present typical features of a HTM: (1) delocalized highest occupied molecular orbital (HOMO); (2) hole reorganization energies higher than the electronic ones; and (3) transparency in the visible region of the electromagnetic spectrum. Among the investigated compounds, 4-(4-phenyl-4-α-naphthylbutadienyl)-N,N-di(4-tolyl)-phenylamine (HTM1) shows the most promising features in terms of optimal charge delocalization, high electronic couplings, and low energetic disorder that cause a high hole mobility in the amorphous phase. The obtained results are in qualitative and quantitative agreement with the experimental data and suggest that the current computational approach could be further employed to obtain valuable insights for the design of new HTMs aiming at improving the performances of presently available OSCs.
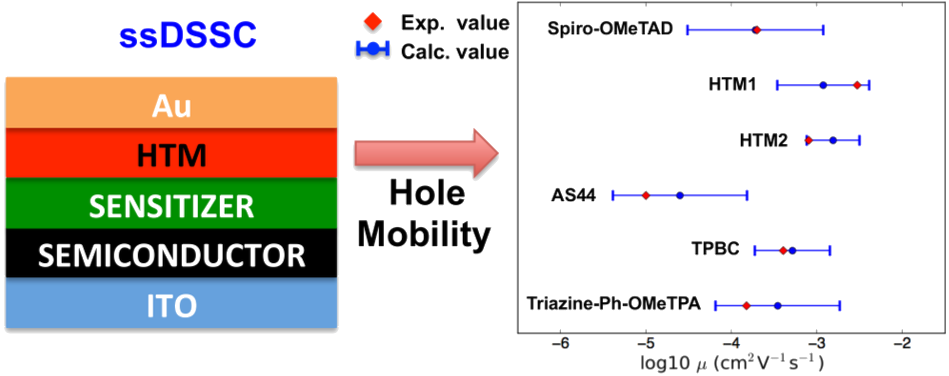
Figure 1: Schematic representation of a ssDSSC and hole mobility calculated for the six HTMs studied.
[1] D. Alberga et al., Phys. Chem. Chem. Phys. 17 (28), 18742-18750 (2015).
[2] D. Alberga et al., J. Phys. Chem. C 19 (42), 23890-23898 (2015).
Contributed talk
Multifunctional organometallic molecular junctions
1Institute of Chemical Sciences of Rennes, CNRS - University of Rennes 1, France
2School of Material Science and engineering, Nanyang University, Singapore
3Laboratory for Chemistry of Novel Materials, University of Mons, Belgium
Molecular materials based on the assemblage of metallic fragments and conjugated organic or inorganic ligands present additional features compare to pure organic systems. This is due to their relatively good stability combined with active excited-states and redox properties which confer them interesting physical properties in several domains (magnetism, optics, electrochemistry...). These multifunctional systems are promising candidates to be incorporated in nanoscale devices for molecular electronics, opto-electronics and/or spintronics.
The control and manipulation of the physical properties of such systems is the key element for the developments of this research, but it presents considerable conceptual challenges. These challenges can be met by combining experimental and theoretical studies. Using results obtained by quantum chemical calculations, mainly of density-functional theory (DFT)-type, associated with experimental measurements, some physical properties of representative compounds can be explained and predicted. Particular emphasis will be put on the electronic reasons of the changes of physical properties upon oxidation or reduction, or upon structure modifications induced by irradiation or temperatue. This will be illustrated by detailing recent studies performed in our groups briefly explained in Figure 1 [1,2].
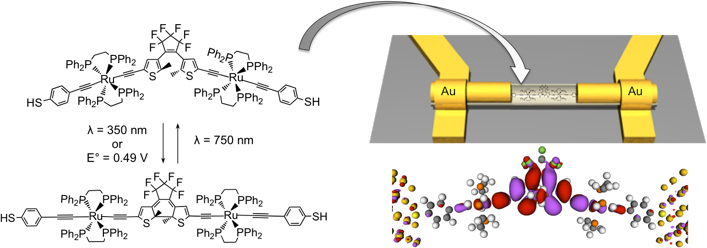
Figure 1: Representation of the photo- and electro-chromic system (left) which was immobilized in gold-gold junction (top right). Conductivity was studied experimentally and theoretically (NEGF formalism, bottom right: molecular projected spectrum Hamiltonian at the Fermi level represented at zero-bias).
[1] C. Lagrost, K. Costuas, N. Tchouar, H. Le Bozec, S. Rigaut, Chem. Commun., 6117 (2008).
[2] F. Meng, Y.-M. Hervault, L. Norel, K. Costuas, C. Van Dyck, V. Geskin, J. Cornil, H. H. Hng, S. Rigaut, X. Chen, Chem. Sci. 3, 3113 (2012).
Invited talk
Simulation of electron transport in molecular junctions using multiconfiguration wavefunction and reduced density matrix methods
Department of Physics, University of Erlangen-Nuremberg, Germany
Nonequilibrium electron transport in molecular junctions often involves correlation effects due to electron-electron or electron-vibrational interaction. In this talk, methods are discussed, which allow an accurate description of correlated electron transport, including the hierarchical master equation approach and the multilayer multiconfiguration time-dependent Hartree (ML-MCTDH) method [1]. Moreover, the combination of the ML-MCTDH method with reduced density matrix theory is outlined [2]. The performance of the methods is discussed based on models for vibrationally coupled electron transport in molecular junctions, including both time-dependent and steady-state transport.
[1] H. Wang and M. Thoss, J. Chem. Phys. 131, 024114 (2009); 138, 134704 (2013).
[2] E. Wilner, H. Wang, G. Cohen, M. Thoss, E. Rabani, Phys. Rev. B 88, 045137 (2013); 92, 195143 (2015).
Contributed talk
Current induced phonons renormalization in molecular junctions
1Key Laboratory for the Physics and Chemistry of Nanodevices, Department of Electronics, Peking University, Beijing 100871, China
2School of Physics, AMBER and CRANN Institute, Trinity College, Dublin 2, Ireland
3Materials Division, National Physical Laboratory, Teddington, TW11 0LW, United Kingdom
4Beida Information Research (BIR), Tianjin 300457, China
Current-induced forces are at the origin of a rich variety of effects, including vibrations, rotations as well as mass and energy flow at interfaces [1]. We explain how the electrical current flow in a molecular junction can modify the vibrational spectrum of the molecule by renormalizing its normal modes of oscillations.
This is demonstrated with first-principles self-consistent transport theory, where the current-induced forces are evaluated from the expectation value of the ionic momentum operator.
The redistribution of the electron density caused by the presence of a stationary current – causing atomic rearrangements and modifying the instantaneous Hamiltonian – is calculated using the non-equilibrium Green's function formalism combined with Density Functional Theory.
We explore here the case of H2 sandwiched between two Au electrodes and show that the current produces stiffening of the transverse translational and rotational modes and softening of the stretching one along the current direction. Such behavior is understood in terms of charge redistribution, potential drop and elasticity changes as a function of the current.
[1] T. N. Todorov et al, Eur. J. Phys. 35, 065004 (2014).
[2] Meilin Bai, Clotilde S. Cucinotta, Zhuoling Jiang, Ivan Rungger, Stefano Sanvito, and Shimin Hou, Current induced phonons renormalization in molecular junctions, under review.